#!/usr/bin/env python3 # -*- coding: utf-8 -*- """ Created on Tue Jul 12 18:38:35 2022 https://janaf.nist.gov/tables/H-083.html #Database Symmetry Number: Table 10.1 and Appendix B of C. Cramer “Essentials of Computational Chemistry”, 2nd Ed. spin: 0 for molecules in which all electrons are paired, 0.5 for a free radical with a single unpaired electron, 1.0 for a triplet with two unpaired electrons, such as O_2. @author: qli """ import numpy as np from ase.io import read, write from ase.thermochemistry import IdealGasThermo from scipy import constants as con
atoms = read('./freq/POSCAR') sym = 3# symmetry number of NH3 spin = 0# spin of NH3. tem = 298.15# Temperature out = read('./OUTCAR', format='vasp-out') potentialenergy = out.get_potential_energy()
model = read('./freq/POSCAR') # model_positions = model.get_positions()
vib_energies = [] withopen('./freq/OUTCAR') as f_in: lines = f_in.readlines() for num, line inenumerate(lines): if'cm-1'in line: vib_e = float(line.rstrip().split()[-2]) vib_energies.append(vib_e)
vib_energies = np.array(vib_energies[:-6])/1000# For Gas, the last six are translation and rotation # zpe = sum(vib_energies)/2
qli@bigbro test_nh3 % ls CONTCAR INCAR OUTCAR freq/ H-083.txt KPOINTS POSCAR get_gas_entropy.py* qli@bigbro test_nh3 % python3 get_gas_entropy.py Entropy components at T = 298.15 K and P = 101325.0 Pa: ================================================= S T*S S_trans (1 bar) 0.0014947 eV/K 0.446 eV S_rot 0.0004981 eV/K 0.149 eV S_elec 0.0000000 eV/K 0.000 eV S_vib 0.0000047 eV/K 0.001 eV S (1 bar -> P) -0.0000011 eV/K -0.000 eV ------------------------------------------------- S 0.0019964 eV/K 0.595 eV ================================================= G -19.220631915628093 S 192.6230402411749 J/K/mol
虽然书名叫The hard Way,这本书还是给了很多人一种DFT极品快餐的感觉;更会越来越”毁了”专业做计算的,让各种做实验的做计算的扰乱市场。虽然有这种负面的作用,但我还是想尽自己可能,稍微提高我们做计算的平均水平。因此,认真声明一下:本教程的初衷是给专门做计算菜鸟准备的。对于那些以为靠本书就可以快速入门的筒子们(不管是做计算还是做实验的),劝你们还是趁早放弃本书的学习,科研的路上,从来就没有速成二字。
在BB了这么多废话之后,感觉时候再强调下本书的重点了:The Hard Way。The Hard Way 在本人的眼里,类似于书山有路勤为径,学海无涯苦作舟的感觉,类似于经过无数瞎折腾终于尝到一点点小胜利甜头的感觉,类似于苦尽甘来,柳暗花明又一村的感觉。所以:Hard Way是本书的重点,也是痛苦学习的一个过程。别人耗费精力和时间开发出来的软件,短时间就精通是不可能的事情。所以,也别指望着一口气吃成个胖子,耐住寂寞。
本人学习和使用VASP已经快10年了,该教程差不多也5岁了。我进入课题组之前,不会用Ubuntu系统,也不会用脚本,也不会用python写脚本,也没用过VASP。我的VASP入门,是花了一个月看了Davide Sholl的书后,老板又手把手花了一个多月的时间学会的,学习的资料是老版本的Hand on Session系列。每天做的是系列中的练习,准备老板布置的作业,思考老板预留的问题,然后第二天听老板解释每个参数的含义,里面的关键点。而真正操作起来进入课题的时候,又有组里细心的博后专门帮忙指导答疑,也有很多人可以进行讨论学习。写这么多并不是在秀自己遇到了一个多好的老板,多好的课题组。在这样的环境中,经过了四年的学习,好几个课题的实战,才变成你们感觉很牛逼的大师兄(其实我只是经验比你们多一点点而已)。而这本书,也是本人这四年来学习到的精华,每一节都要花费差不多4个小时的样子才能完成,或者更多。而这四年的精华,岂能让你把这本书看一遍就掌握了? 到现在为止,瞎BB了76节。每节你花一天的时间去学习,加上其他参考书的学习,也需要2个月的时间。而2个月,对一个新手来说,完全在承受范围之内,请相信时间的力量。这里,我们澄清下对新手的定义:全职搞计算的菜鸟。所以,Hard Way 就是需要你花掉每一个整天的时间,一点点认真琢磨,反复操练,绝对不是什么速成。而现在我们搞科研都很浮躁,什么都喜欢图快,我们也有句古话:欲速则不达。因此,打算朝夕之内就学会VASP的读者,本书可能真的不适合你。
虽然书名叫The hard Way,这本书还是给了很多人一种DFT极品快餐的感觉;更会越来越”毁了”专业做计算的,让各种做实验的做计算的扰乱市场。虽然有这种负面的作用,但我还是想尽自己可能,稍微提高我们做计算的平均水平。因此,认真声明一下:本教程的初衷是给专门做计算菜鸟准备的。对于那些以为靠本书就可以快速入门的筒子们(不管是做计算还是做实验的),劝你们还是趁早放弃本书的学习,科研的路上,从来就没有速成二字。
在BB了这么多废话之后,感觉时候再强调下本书的重点了:The Hard Way. The Hard Way 在本人的眼里,类似于书山有路勤为径,学海无涯苦作舟的感觉,类似于经过无数瞎折腾终于尝到一点点小胜利甜头的感觉,类似于苦尽甘来,柳暗花明又一村的感觉。所以:Hard Way是本书的重点,也是痛苦学习的一个过程。别人耗费精力和时间开发出来的软件,短时间就精通是不可能的事情。所以,也别指望着一口气吃成个胖子,耐住寂寞。
本人学习和使用VASP已经快10年了,该教程差不多也5岁了。我进入课题组之前,不会用Ubuntu系统,也不会用脚本,也不会用python写脚本,也没用过VASP。我的VASP入门,是花了一个月看了Davide Sholl的书后,老板又手把手花了一个多月的时间学会的,学习的资料是老版本的Hand on Session系列。每天做的是系列中的练习,准备老板布置的作业,思考老板预留的问题,然后第二天听老板解释每个参数的含义,里面的关键点。而真正操作起来进入课题的时候,又有组里细心的博后专门帮忙指导答疑,也有很多人可以进行讨论学习。写这么多并不是在秀自己遇到了一个多好的老板,多好的课题组。在这样的环境中,经过了四年的学习,好几个课题的实战,才变成你们感觉很牛逼的大师兄(其实我只是经验比你们多一点点而已)。而这本书,也是本人这四年来学习到的精华,每一节都要花费差不多4个小时的样子才能完成,或者更多。而这四年的精华,岂能让你把这本书看一遍就掌握了? 到现在为止,瞎BB了76节。每节你花一天的时间去学习,加上其他参考书的学习,也需要2个月的时间。而2个月,对一个新手来说,完全在承受范围之内,请相信时间的力量。这里,我们澄清下对新手的定义:全职搞计算的菜鸟。所以,Hard Way 就是需要你花掉每一个整天的时间,一点点认真琢磨,反复操练,绝对不是什么速成。而现在我们搞科研都很浮躁,什么都喜欢图快,我们也有句古话:欲速则不达。因此,打算朝夕之内就学会VASP的读者,本书可能真的不适合你。
#!/usr/bin/env python3 # -*- coding: utf-8 -*- """ @author: qli Created on Thu Jun 2 22:09:18 2022 Creat POSCAR for improved dimer calculations from frequency calculations. 1) run frequency calculation: IBRION = 5 POTIM = 0.015 NFREE = 2 NWRITE = 3 ### Must be 3 2) run this script: get_dimer.py 3) use the POSCAR for IDM calc. NSW = 100 Prec=Normal IBRION=44 ! use the dimer method as optimization engine EDIFFG=-0.05 POTIM = 0.05 """
import numpy as np from ase.io import read, write import os from sys import exit
model = read('POSCAR_relax') ### POSCAR_relax is the POSCAR before freq calculations, that means some atoms are not fixed. #model_positions = model.get_positions() model.write('POSCAR_dimer', vasp5=True)
# print(model_positions) # print(len(model))
l_start = 0 # the number of line which contains 'Eigenvectors after division by SQRT(mass)' with open('OUTCAR') as f_in: lines = f_in.readlines() for num, line in enumerate(lines): if 'Eigenvectors after division by SQRT(mass)' in line: l_start = num
if l_start == 0: print('''Check Frequency results and then rerun this script.\n**Remember**: NWRITE must be 3. BYEBYE! ''' ) exit() freq_infor_block = lines[l_start:] l_position = 0 wave_num = 0.0 for num, line in enumerate(freq_infor_block): if 'f/i' in line: wave_tem = float(line.rstrip().split()[6]) if wave_tem > wave_num: wave_num = wave_tem l_position = num+2
vib_lines = freq_infor_block[l_position:l_position+len(model)] for line in vib_lines: infor = line.rstrip().split()[3:] pos_dimer.write(' '.join(infor)+'\n')
pos_dimer.close() print(''' DONE! Output file is named as: POSCAR_dimer and can be used for dimer calculations. Don't forget to rename POSCAR_dimer to POSCAR before you run the dimer jobs. ''')
In VTST v2.04 and later, a modification is required in main.F for the solid-state NEB.
Find and replace:
CALL CHAIN_FORCE(T_INFO%NIONS,DYN%POSION,TOTEN,TIFOR, & LATT_CUR%A,LATT_CUR%B,IO%IU6) with
CALL CHAIN_FORCE(T_INFO%NIONS,DYN%POSION,TOTEN,TIFOR, & TSIF,LATT_CUR%A,LATT_CUR%B,IO%IU6) For vasp.6.2 also find and replace:
IF (LCHAIN) CALL chain_init( T_INFO, IO) with
CALL chain_init( T_INFO, IO) For vasp.6.2.1 you will need vtstcode 4.1 (revision 182). A version of the vtstcode that will work with vasp.6.1.x - vasp.6.2.0 has been saved in the vtstcode6.1 directory.
To build the code, the VASP makefile needs to be changed. Find the variable SOURCE, which defines which objects will be built, and add the following objects before chain.o:
bfgs.o dynmat.o instanton.o lbfgs.o sd.o cg.o dimer.o bbm.o \ fire.o lanczos.o neb.o qm.o opt.o The objects dynmat.o, neb.o, dimer.o, lanczos.o, and instanton.o must be in the SOURCE list before chain.o appears. The optimizer objects, sd.o, cg.o, qm.o, lbfgs.o, bfgs.o, and fire.o must appear before the optimizer driver opt.o. Nothing else needs to be done.
官网里面有句话:Find the variable SOURCE, which defines which objects will be built, and add the following objects before chain.o。
# -*- coding: utf-8 -*- """ Qiang Li Command: python3 vib_correct.py This is a temporary script file. """ import numpy as np from ase.io import read, write import os
#获取虚频开始行 l_position = 0#虚频振动方向部分在OUTCAR中的起始行数 withopen('OUTCAR') as f_in: lines = f_in.readlines() wave_num = 0.0 for num, line inenumerate(lines): if'f/i'in line: wave_tem = float(line.rstrip().split()[6]) if wave_tem > wave_num: #获取最大的虚频 wave_num = wave_tem l_position = num+2 # ASE读POSCAR model = read('POSCAR') model_positions = model.get_positions() num_atoms = len(model) #print(model_positions)
# 获取虚频对应的OUTCAR部分 vib_lines = lines[l_position:l_position + num_atoms] #振动部分 7222到7248行 #print(vib_lines) vib_dis = [] for line in vib_lines: #model_positions = [float(i) for i in line.rstrip().split()[:3]] vib_infor = [float(i) for i in line.rstrip().split()[3:]] # dx, dy, dz对应的那三列 vib_dis.append(vib_infor) vib_dis = np.array(vib_dis) #将振动部分写到一个array中。
#!/usr/bin/env python # -*- coding: utf-8 -*- """ Qiang Li Command: python3 vib_correct.py This is a temporary script file. """ import numpy as np import os
#获取虚频开始行 l_position = 0#虚频振动方向部分在OUTCAR中的起始行数 withopen('OUTCAR') as f_in: lines = f_in.readlines() wave_num = 0.0 for num, line inenumerate(lines): if'f/i'in line: wave_tem = float(line.rstrip().split()[6]) if wave_tem > wave_num: #获取最大的虚频 wave_num = wave_tem l_position = num+2 # 读POSCAR withopen('POSCAR') as f_pos: lines_pos = f_pos.readlines()
# 获取虚频对应的OUTCAR部分 num_atoms = sum([int(i) for i in lines_pos[6].rstrip().split()]) vib_lines = lines[l_position:l_position + num_atoms] #振动部分 7222到7248行
model_positions = [] vib_dis = [] for line in vib_lines: position = [float(i) for i in line.rstrip().split()[:3]] vib_infor = [float(i) for i in line.rstrip().split()[3:]] # dx, dy, dz对应的那三列 model_positions.append(position) vib_dis.append(vib_infor)
###保存结构 f_out = open('POSCAR_new','w') f_out.writelines(lines_pos[:8]) f_out.write('Cartesian\n') for i in new_positions: f_out.write(' '.join([str(coord) for coord in i]) + ' F F F\n') f_out.close()
import numpy as np from ase.io import read, write import os, sys
''' Important Note: Atoms are indexed from 0. ''' atom_A, atom_B = [int(i) for i in sys.argv[1:3]] model = read('POSCAR') positions = model.get_positions() dis_AB_1 = model.get_distance(atom_A, atom_B, mic=True) dis_AB_2 = model.get_distance(atom_A, atom_B, mic=False)
print(round(dis_AB_1, 4), '\t',round(dis_AB_2,4))
## Not Used #coord_A = positions[atom_A] #coord_B = positions[atom_B] #dis_AB = np.linalg.norm(coord_A-coord_B) #print(round(dis_AB,4))
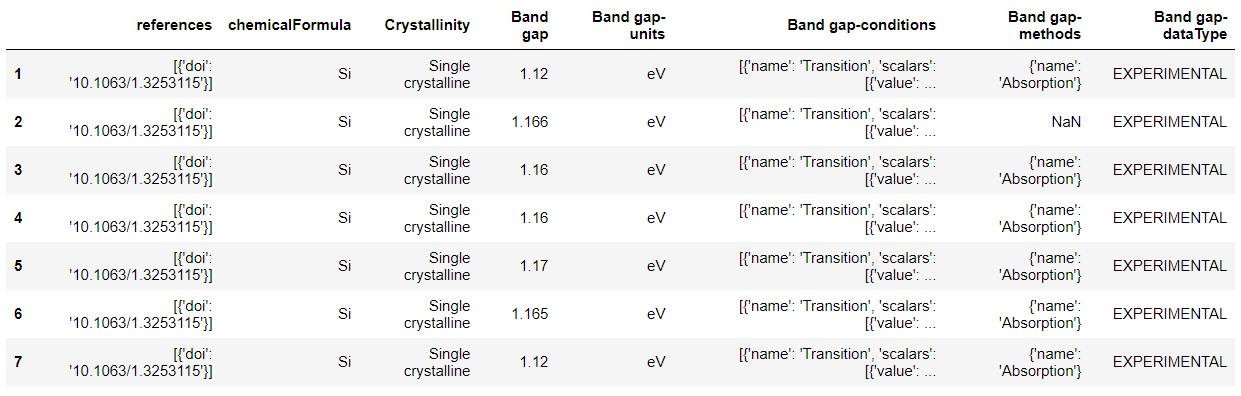
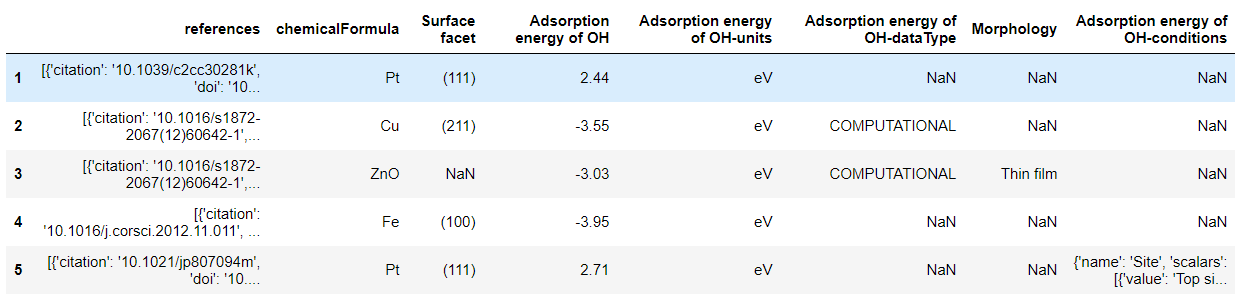
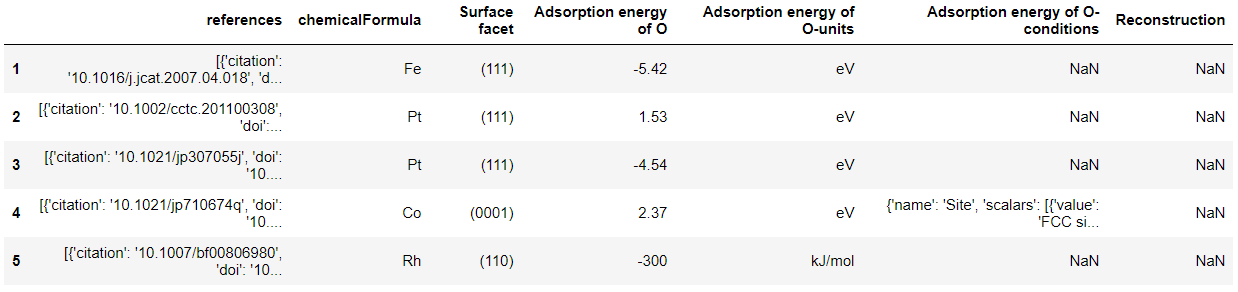
# 我们发现这里不需要指定筛选条件,只需要返回所需要的性质即可。 df_OH = cdr.get_dataframe(criteria={}, properties=['adsorption energy of OH'], secondary_fields=True) df_O = cdr.get_dataframe(criteria={}, properties=['adsorption energy of O'], secondary_fields=True)
0%| | 0/9 [00:00<?, ?it/s]d:\programs\anaconda3\envs\py36\lib\site-packages\matminer\data_retrieval\retrieve_Citrine.py:103: FutureWarning:
pandas.io.json.json_normalize is deprecated, use pandas.json_normalize instead
100%|███████████████████████████████████████████████████████████████████████████████████| 9/9 [00:00<00:00, 128.63it/s]
all available fields:
['uid', 'Adsorption energy of OH', 'Adsorption energy of OH-units', 'Morphology', 'Surface facet', 'category', 'references', 'chemicalFormula', 'Adsorption energy of OH-conditions', 'Adsorption energy of OH-dataType']
suggested common fields:
['references', 'chemicalFormula', 'Surface facet', 'Adsorption energy of OH', 'Adsorption energy of OH-units', 'Adsorption energy of OH-dataType', 'Morphology', 'Adsorption energy of OH-conditions']
100%|█████████████████████████████████████████████████████████████████████████████████| 21/21 [00:00<00:00, 140.70it/s]
all available fields:
['uid', 'Surface facet', 'Adsorption energy of O', 'category', 'references', 'chemicalFormula', 'Reconstruction', 'Adsorption energy of O-units', 'Adsorption energy of O-conditions']
suggested common fields:
['references', 'chemicalFormula', 'Surface facet', 'Adsorption energy of O', 'Adsorption energy of O-units', 'Adsorption energy of O-conditions', 'Reconstruction']
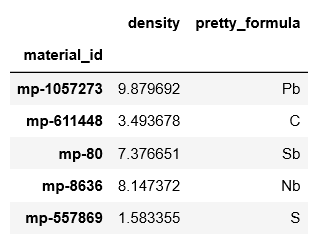
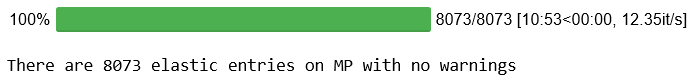
''' 除了上次的搜索条件外,我们想搜索包含 Pb 和 Te 的材料:"elements": {"$all": ["Pb", "Te"]} 材料的稳定性也在考虑之中,energy above hull 必须在1e-6以下:"e_above_hull": {"$lt": 1e-6} ''' df = mpdr.get_dataframe(criteria={"elasticity": {"$exists": True}, "elasticity.warnings": [], "elements": {"$all": ["Pb", "Te"]}, "e_above_hull": {"$lt": 1e-6}}, # to limit the number of hits for the sake of time properties = ["elasticity.K_VRH", "elasticity.G_VRH", "pretty_formula", "e_above_hull", "bandstructure", "dos"])
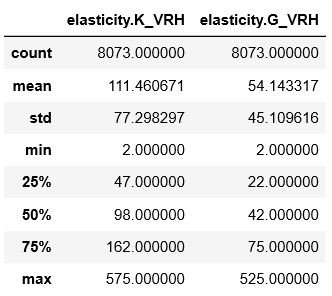
print("There are {} elastic entries on MP with no warnings that contain " "Pb and Te with energy above hull ~ 0.0 eV".format(df['elasticity.K_VRH'].count()))
结果是:
There are 3 elastic entries on MP with no warnings that contain Pb and Te with energy above hull ~ 0.0 eV
1
df.head()
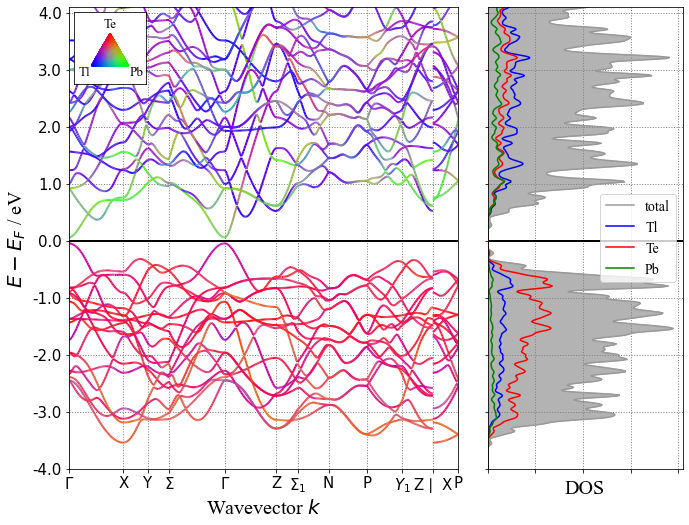
下面来查看一下其中一个材料的能带和DOS图
1 2 3 4 5 6
from pymatgen.electronic_structure.plotter import BSDOSPlotter # 调用 Pymatgen 作图
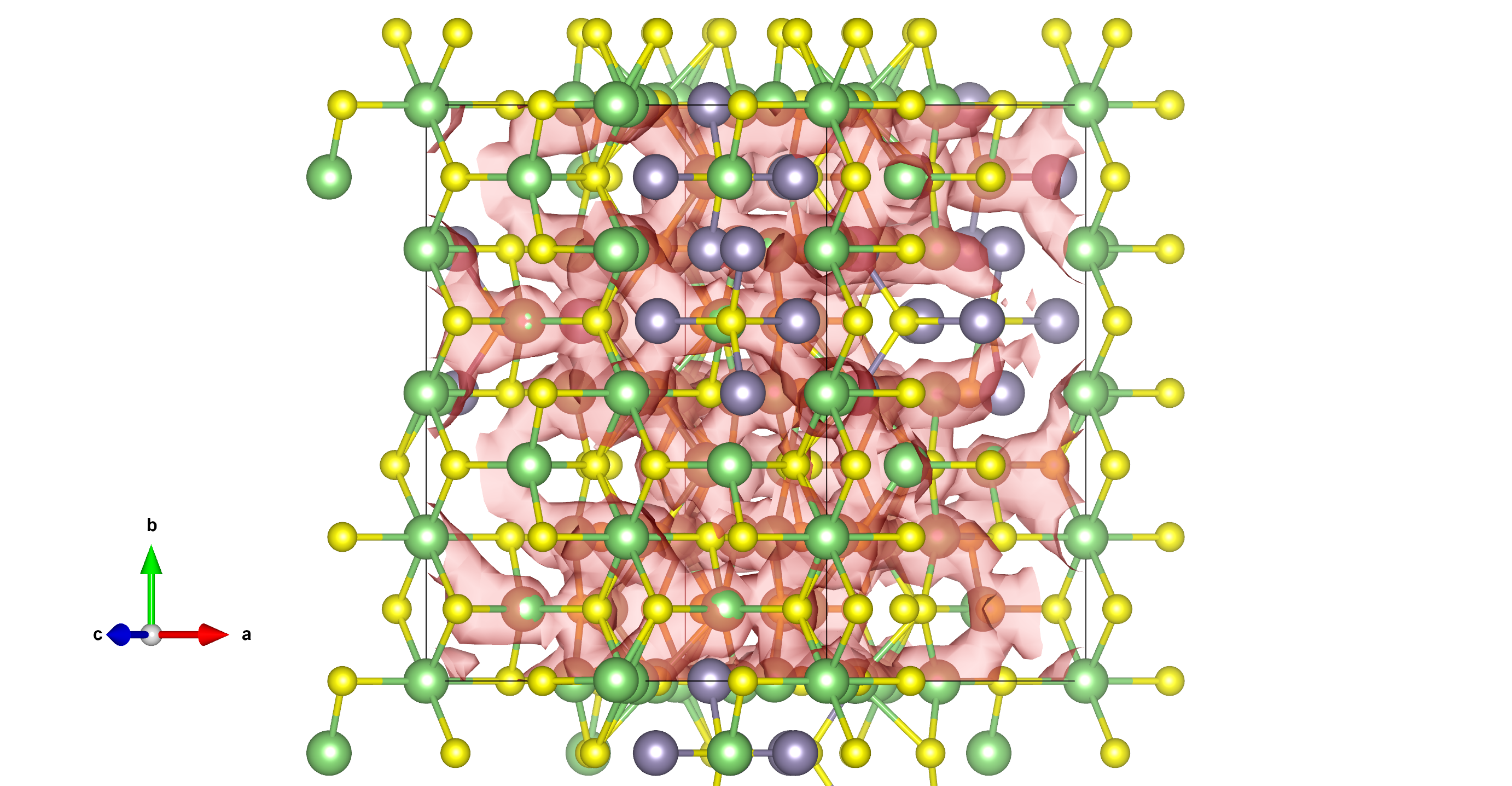
作者总结说 “A general principle for the design of Li-ion conductors with low activation energy can be distilled from the above findings: all of the sites within the diffusion network should be energetically close to equivalent, with large channels connecting them.”
我们把其中的 pymatgen_diffusion 文件夹放到 Anaconda的site-packages文件夹下,路径是 Windows 系统:……\Anaconda\Lib\site-packages;Linux系统:……/anaconda3/lib/pythonx.x/site-packages,就算安装好了。
接下来我们可以启动python,导入这个模块,如果不报错就没有问题了。
1 2 3 4 5 6
[test@ln0%tianhe2 li_sn_s]$ python Python 3.8.3 (default, Jul 2 2020, 16:21:59) [GCC 7.3.0] :: Anaconda, Inc. on linux Type "help", "copyright", "credits" or "license" for more information. >>> import pymatgen_diffusion >>>
学习用法
我们可以在其github网站上通过例子学习这个模块的用法。
点击打开 probbility_analysis.ipynb 文件。
其内容如下(有所删减):如果不想看的话可直接查看 开始作图 部分
1 2 3 4 5 6 7 8 9 10 11 12 13 14
from pymatgen.analysis.diffusion_analyzer import DiffusionAnalyzer from pymatgen_diffusion.aimd.pathway import ProbabilityDensityAnalysis import json
data = json.load(open("../pymatgen_diffusion/aimd/tests/cNa3PS4_pda.json", "r")) diff_analyzer = DiffusionAnalyzer.from_dict(data) # 初始化DiffusionAnalyzer类
pda = ProbabilityDensityAnalysis.from_diffusion_analyzer(diff_analyzer, interval=0.5, species=("Na", "Li")) #可以指定离子 #Save probability distribution to a CHGCAR-like file pda.to_chgcar(filename="CHGCAR_new2.vasp") #保存概率密度文件
开始作图
代码(test.py)如下:
1 2 3 4 5 6 7 8 9
from pymatgen_diffusion.aimd.pathway import ProbabilityDensityAnalysis from pymatgen.core.trajectory import Trajectory from pymatgen.io.vasp.outputs import Xdatcar from pymatgen.analysis.diffusion_analyzer import DiffusionAnalyzer
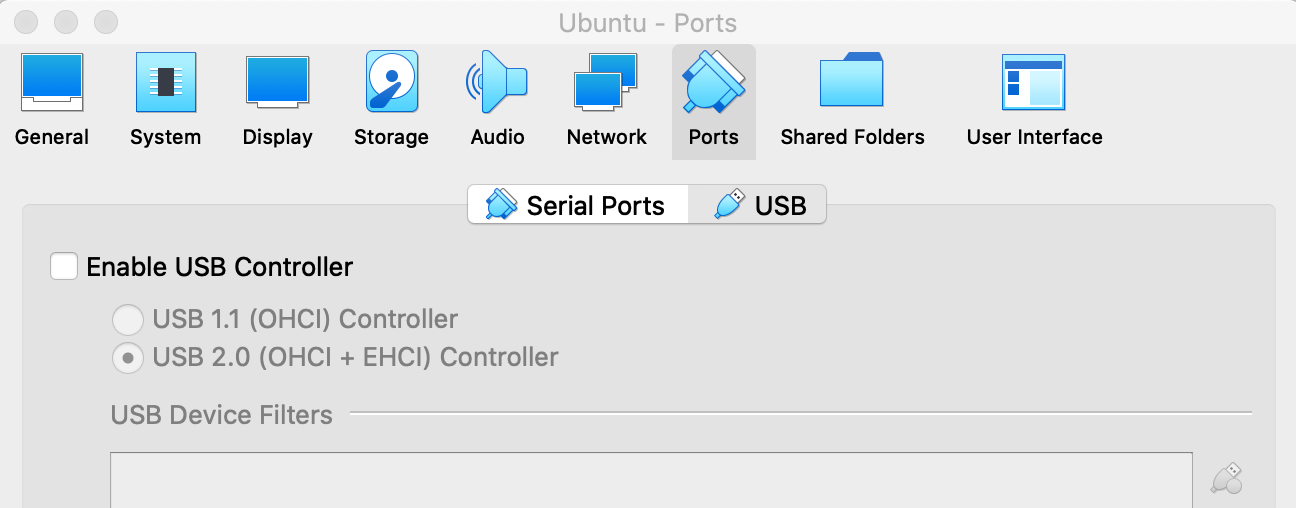
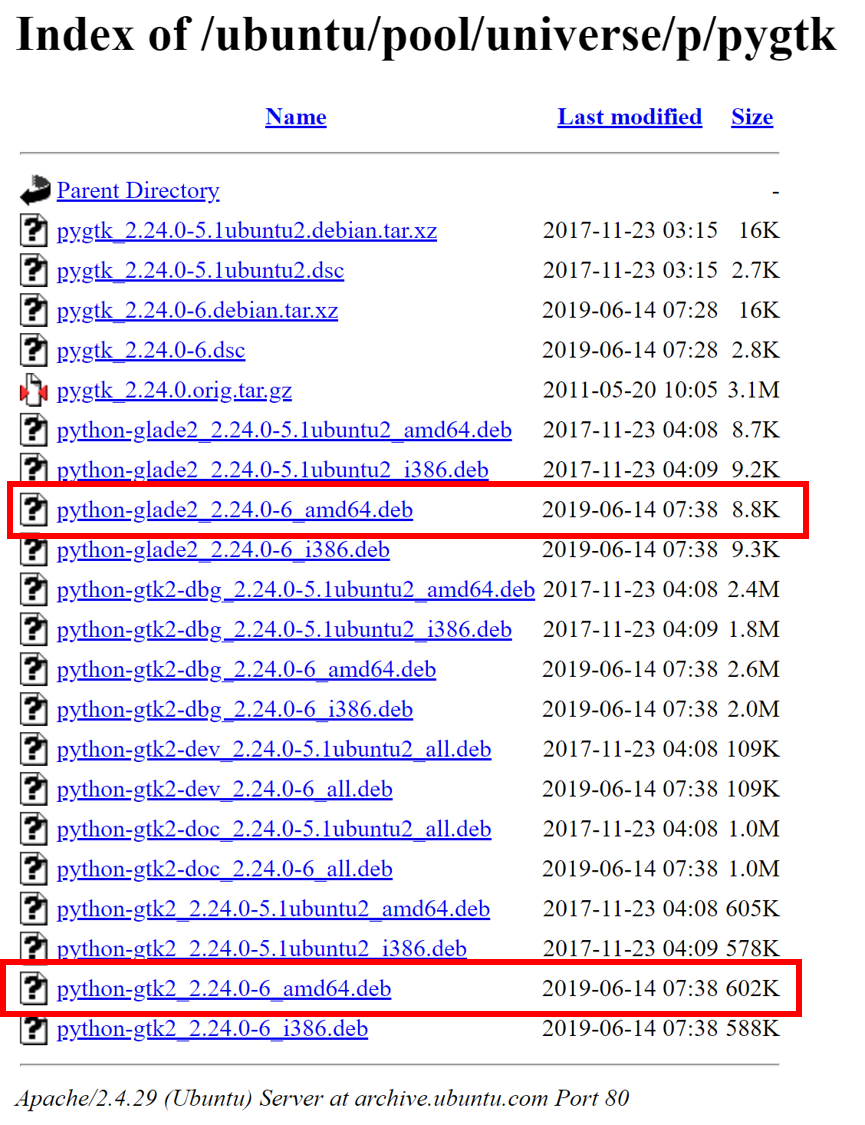
(base) qli@bigbrosci:~$ p4v File "/usr/bin/p4v.py", line 9 print"""You need to get version 2.0 (or later) of PyGTK for this to work. You can get source code from http://www.pygtk.org """ ^ SyntaxError: invalid syntax (base) qli@bigbrosci:~$ which p4v /usr/bin/p4v (base) qli@bigbrosci:~$ sudo vi /usr/bin/p4v [sudo] password for qli: (base) qli@bigbrosci:~$ sudo vi /usr/bin/p4v.py (base) qli@bigbrosci:~$ grep python /usr/bin/p4v export PYTHONPATH=$PYTHONPATH:/usr/lib/python2.7/dist-packages exec python2 /usr/bin/p4v.py "$@" (base) qli@bigbrosci:~$ grep python /usr/bin/p4v.py #!/usr/bin/env python2
#!/usr/bin/env python3 # -*- coding: utf-8 -*- """ convert SMILES to xyz """ import sys from openbabel import pybel import numpy as np import ase import ase.io from ase import Atoms from ase.constraints import FixAtoms smiles, out_name = sys.argv[1:3]
''' Introduction to SMILES: https://en.wikipedia.org/wiki/Simplified_molecular-input_line-entry_system https://www.daylight.com/dayhtml/doc/theory/theory.smiles.html '''
## Use openbabel to convert SMILES to xyz. mol = pybel.readstring("smi", smiles) mol.make3D(forcefield='mmff94', steps=100) # mol.write("xyz", filename=out_name+'_pybel.xyz', overwrite=True)
# USE ase to make POSCAR ''' https://wiki.fysik.dtu.dk/ase/ase/atoms.html''' geo = Atoms() geo.set_cell(np.array([[16.0, 0.0, 0.0],[0.0, 17.0, 0.0],[0.0, 0.0, 18.0]])) geo.set_pbc((True,True,True)) for atom in mol: atom_type = atom.atomicnum atom_position = np.array([float(i) for i in atom.coords]) geo.append(atom_type) geo.positions[-1] = atom_position geo.center()
'''https://wiki.fysik.dtu.dk/ase/ase/constraints.html''' c = FixAtoms(indices=[atom.index for atom in geo if atom.symbol == 'XX']) geo.set_constraint(c)
1 geo = Atoms() 2 geo.set_cell(np.array([[16.0, 0.0, 0.0],[0.0, 17.0, 0.0],[0.0, 0.0, 18.0]])) 3 geo.set_pbc((True,True,True)) 4for atom in mol: 5 atom_type = atom.atomicnum 6 atom_position = np.array([float(i) for i in atom.coords]) 7 geo.append(atom_type) 8 geo.positions[-1] = atom_position 9 geo.center()
'''https://wiki.fysik.dtu.dk/ase/ase/constraints.html''' 10 c = FixAtoms(indices=[atom.index for atom in geo if atom.symbol == 'XX']) 11 geo.set_constraint(c)