Ex24 乙醇分子的振动频率计算(二)
按照前面一节介绍的方法,结构优化过程完毕后,准备频率计算的输入文件,提交任务等待结束。然后我们介绍一下频率计算的输出与POSCAR原子的固定。
1 查看结果
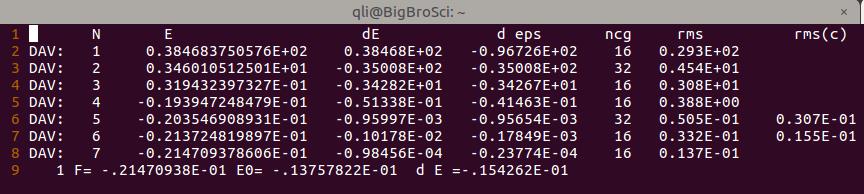
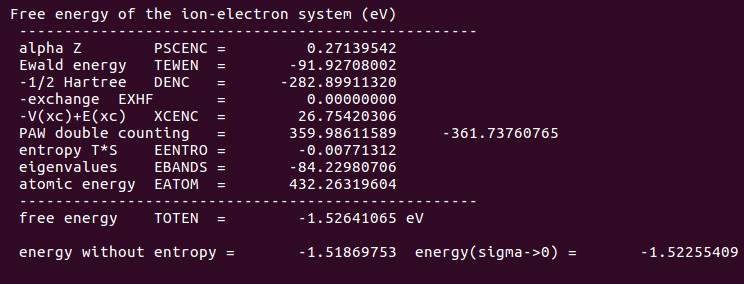
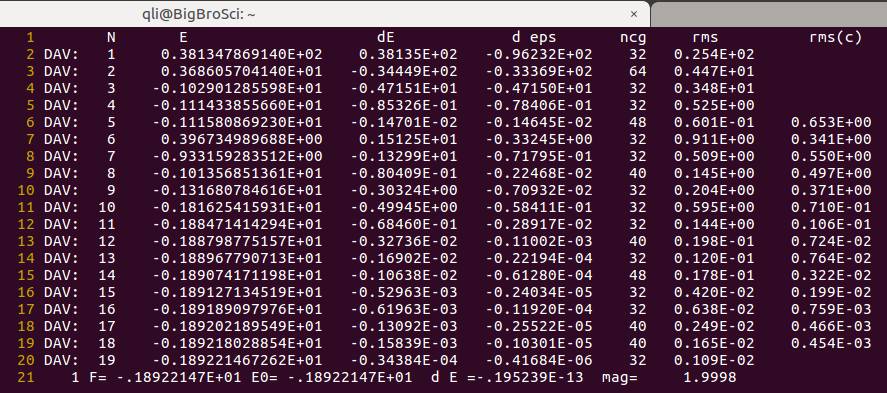
1.1 查看OSZICAR,你会发现一共计算了55步。

下面我们分析一下这$55$步是怎么回事:
A)乙醇分子$\rm{CH{3}CH{2}OH}$ 含有 $9$ 个原子,每个原子在 $x, y, z$三个方向上均有一个自由度,共 $9 \times 3 = 27$ 个
B)我们设置的NFREE=2 ,也就是在每个方向上 +POTIM 和 –POTIM都移动并算一下,这样就有了 $27 \times 2 = 54$ 步:官网原文如下:大家自己去查阅IBRION和NFREE的相关内容。
The parameter NFREEdetermines how many displacements are used for each direction and ion, andPOTIM determines the step size. The step size is defaulted to 0.015 ? (startingfrom VASP.5.1), if too large values are supplied in the input file. Expertiseshows that this is a very reasonable compromise.`
NFREE=2 usescentral differences, i.e., each ion is displaced by a small positive andnegative displacement, ±POTIM, along each of the cartesian directions.`
C)还有一步:$55 -54 = 1$, 这一步指的是第一个离子步,为频率计算前的单点计算。
所以当你设置了NFREE=2的时候,频率计算需要 $1+N \times 6$ 步。$N$ 为体系中的振动的原子数。
2 固定原子
默认的是所有的原子在$x, y, z$三个方向上均可移动。但很多时候,我们只需要振动感兴趣的原子或者某一特定的方向就可以了,也就是说选择性计算频率。比如我们只想算乙醇的羟基振动峰,那么其他的原子就可以固定。VASP可以通过设置POSCAR来实现这个功能?首先我们看一下当前的POSCAR:

左下角的:set nu 显示文本的行数,取消行数可以通过 :set nonu
如果想要选择性地固定某些原子,我们需要以下几个步骤:
1) 在第七行和第8行之间插入一行,内容为 Selective Dynamics ,前面讲过 VASP 只认第一个字母,也就是S是必须的,Selective Dynamics 和 See, Sea….等其他S开头的都是一个效果的。
2) 加入Selective之后,我们需要在每一行的坐标后面加上 T 或者F表示允许和禁止移动。这里我们需要加三个T或者F,表示在 $x, y, z$ 三个方向上选择性固定原子的移动,
三个方向都允许:T T T
三个方向上都不允许: F F F
$x$ 移动,$y$ 和 $z$ 方向上固定为: T F F
$x$ 和 $y$ 方向上固定,$z$ 方向振动: F F T
以此类推,其他的大家根据自己的情况固定。
师兄,那么我们要做的就是在POSCAR中逐行写上T T T或者F F F就行了吧?
是的,下面大师兄教给你的是通过 Vim 的命令或者 p4vasp 实现这个功能。
2.1 通过 Vim 实现原子的固定和选择
第一步:加入 Select 的关键字母S,并在坐标后面全部加上 T T T

图中箭头1:插入一行,告诉 VASP 我们要选择性的固定某些原子或者在某些方向上;
图中方块2 :10,18s 中 s 代表替换(substitute)的意思,这里表示我们选中了第 $10$ 到 $18$ 行,$10$ 和 $18$ 之间有个逗号表示连续;10,18s后面用一个 / 分开,紧跟着你我们要替换的内容;
$ 在这里是末尾的意思,$/T T T我们要把每一行的最后替换成 T T T
后面再用一个/分开,加上g 表示 global 全部替换的意思。
输入完毕后,回车,效果如下:

每一行的末尾都加入了 T T T 箭头指的地方告诉我们:$9$ 行中的 $9$ 个地方发生了替换。
(自己复习下sed的用法,比较两者的区别)
第二步,下面我们要把OH之外的原子全部固定住:
通过 p4vasp 我们可以知道,OH 的两个原子为第一个(O)和最后一个(H),因此我们把第 $11$ 行到 $17$ 行中的所有T替换成F就可以了。我们可以使用 :11,17s/T/F/g来实现。最后效果如下:

C)扩展:
如果所有的原子后面为: T T T,这与我们之前的计算效果是一样的(第 $8$ 行没有S,坐标后面没有T T T)。此外,固定之前要先找到哪些原子我们希望固定的,以及它们在坐标中的顺序。
2.2 使用p4vasp实现上述功能:
基于 p4vasp 的可视化特性,直接用鼠标操作是很多linux 小白最喜欢看到的,下面我们主要讲解一下 p4vasp 的操作,请务必将 Vim 和 p4vasp 的操作关联起来,这样你会就会发现可视化和命令之间的微妙关系了。
1) 打开p4vasp,导入POSCAR;

System 显示 ethanol 说明数据已导入。
2) 鼠标系列操作
A)点一下 Show 按钮,显示乙醇分子结构;
B) 点击Build按钮:显示分子的坐标信息:这里的坐标顺序和POSCAR完全一致:

C) 选择乙醇的羟基(使用空格键选择原子),下图中可以知道 O 和 H 在坐标顺序中为第一个和最后一个;

D)选中右上角的Selective Dynamics,效果如图中箭头2指出来的部分,这个动作和我们之前在第八行插入Selective Dynamics 以及在坐标每一行加入T T T是等效的

E)选中第二行坐标,摁住shift键,然后再点一下倒数第二行,选中整个区域后,点击右上方的 Unselect , 这个动作等效于我们将 $11$ 到 $17$ 行中的T全部替换为F。

F)操作完成后,左上角: File --> Save System As 保存新的POSCAR即可。
4 扩展练习:
4.1 掌握本节的两个POSCAR处理方法;
4.2 学会计算频率分析所需的步数;
4.3 浏览OUTCAR,查找频率输出结果
5 总结:
通过对比手动敲命令修改POSCAR和使用 p4vasp 进行鼠标操作,这里大师兄希望大家能掌握以下三点:
1) 学会 Vim 的使用技巧,当然了,Vim 及其强大,完全掌握基本不可能,但最基本的操作要了解;
2) p4vasp 的操作要熟悉,不知道怎么操作的,导入一个计算文件,随便点点,找找感觉;
3) 在使用 p4vasp 的操作中,你要学会思考:怎么将鼠标操作转化为命令语言来实现,为将来写脚本做好准备,毕竟很多时候,我们用不了可视化界面,就只能手动修改格式了,结合命令,比可视化操作更快,还可以批量进行。